清华新闻网12月18日电 12月12日至14日,第四届医学人工智能大会(CMAIC 2025)在苏州举行。会上,清华大学丘成桐数学科学中心副教授包承龙、2024届博士毕业生郑棣瀚以及求真书院2023级博士生张慧共同提出的“面向蛋白质结构解析的弱配对数据生成算法”入选2025年度医学人工智能代表性算法。

包承龙(左一)、张慧(左二)接受证书颁授
冷冻电子显微镜(Cryo-EM)虽已成为解析生物大分子结构的核心技术,但在实际应用中,由于复杂的成像环境和极低的信噪比,获取高质量的干净-噪声配对训练数据始终是制约AI算法性能的瓶颈。针对这一挑战,包承龙团队构建了一套基于概率图模型的弱配对数据生成理论体系。团队首先提出LUD-VAE模型,在仅有干净域数据x与退化域数据y的边缘样本条件下,用概率图显式拆分结构/内容隐变量z与成像退化/噪声隐变量z_n,并在推断不变性假设下把ELBO写成无需成对样本也可计算的形式,从而学习联合分布并由条件分布p(y|x)把任意干净样本合成逼真的退化观测,批量产出可控的配对训练对。随后,SeNM-VAE面向少量配对+大量不配对的实际情况,采用层级隐变量与混合推断,将仅配对数据、仅干净数据、仅退化数据三类数据统一到同一变分目标中,既保留可解释的退化因子,又显著增强对真实复杂噪声分布的表达能力,生成更贴近实验的配对训练样本。
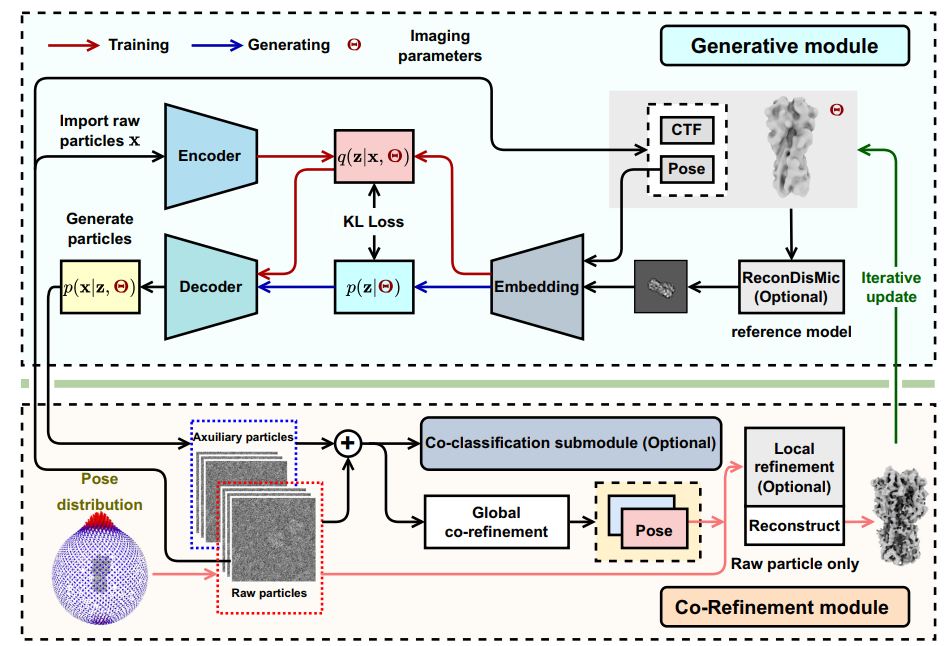
CryoPROS算法架构
在此基础上,团队进一步将弱配对数据生成模型应用于冷冻电镜领域长期存在的“优势取向”难题。由于生物样本易在气液界面以特定方向吸附,导致颗粒在冰层中排列产生严重偏倚,进而引发三维重构失真。团队研发了基于条件层次化变分自编码机(cHVAE)的CryoPROS算法,该算法能在超低信噪比及采样不均衡条件下,高效生成高保真度的辅助颗粒视图。这些生成数据有效补全了实验数据的采样缺口,团队据此提出了生成数据与实验数据联合优化的计算范式,显著降低了颗粒对齐误差并提升了结构解析的稳健性。
目前,该系列成果已在多套实验数据中验证了其有效性,以计算手段突破了传统方法依赖复杂生物样本优化或特殊数据收集策略的局限。CryoPROS算法已被由哈佛大学医学院管理的国际生物软件联盟SBGrid收录。
供稿:丘成桐数学科学中心
编辑:李华山
审核:郭玲