清华新闻网9月4日电 在当今生物医学研究的前沿,单细胞RNA测序(scRNA-seq)技术正引领着一场革命。这项技术能够以前所未有的精度观测单个细胞内的基因活动,帮助我们更好地理解生命系统、疾病原因和生物发育过程。然而,随着单细胞技术的快速发展,生物学家和数据科学家们面临着由海量高维基因数据带来的巨大挑战。
现有的单细胞数据分析方法,包括深度学习算法,往往难以准确捕捉数据中的非线性结构和细微变化,导致重要的生物学信息可能被忽视或误解。特别是在发现罕见的细胞类型、了解细胞如何变化以及绘制完整的“细胞地图”等重要任务中,现有方法的局限性日益凸显。
针对上述问题,清华大学丘成桐数学科学中心丘成桐团队与新加坡国立大学统计与数据科学系姚志刚团队合作,提出一种名为scAMF(Single-Cell Analysis via Manifold Fitting)的创新算法(如图所示)。该算法通过流形拟合进行单细胞分析,旨在从根本上改变分析和理解单细胞数据的方式。scAMF把复杂的单细胞数据看作是由两部分组成的。一部分是有意义的信息结构(低维流形结构),另一部分是无用的干扰信息(环境噪声)。通过流形拟合,scAMF能够有效地提取出潜在的流形结构,保留关键的生物学信息。该方法首先对原始数据进行多种转换,然后利用共享最近邻度量和局部几何结构,自适应地拟合空间结构不同的流形,从而有效处理细胞类型间的复杂非线性关系。经过流形拟合之后,scAMF只需使用简单的聚类方法就可以获得高精度聚类结果。在处理复杂单细胞数据时,scAMF表现优异,在细胞识别的准确性、细胞可视化等方面均优于现有的单细胞分析方法,包括基于图的算法以及深度学习算法。
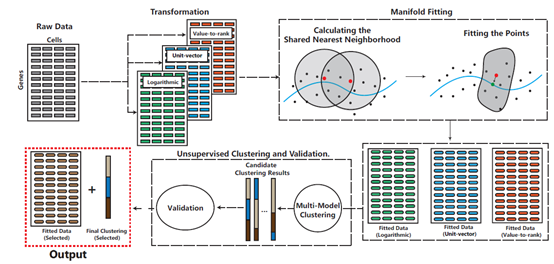
scAMF流程的示意图概述。该过程始于使用三种方法进行数据转换,随后进行流形拟合以去除数据噪声同时保留其结构。拟合后的数据随后经过多种聚类方法,自动地选择最佳聚类结果,最终产生拟合数据和细胞类别作为最终输出
研究团队已经在25个复杂的单细胞数据集上测试了scAMF方法。这些数据集包括人脑细胞的详细信息、胚胎如何随时间发育的数据以及研究癌症多样性的数据等。研究表明,scAMF不仅能够更准确地识别已知细胞类型,还能够提供容易理解且有明确生物学意义的可视化结果。这些发现为理解复杂生命系统的组成和功能提供了新的见解。
9月3日,相关研究成果以“通过流形拟合进行单细胞分析:一种用于RNA聚类及其展望的框架”(Single-Cell Analysis via Manifold Fitting: A Framework for RNA Clustering and Beyond)为题,发表于《美国国家科学院院刊》(Proceedings of the National Academy of Sciences of the United States of America,PNAS)。
新加坡国立大学教授姚志刚为该论文的第一作者,清华大学丘成桐数学科学中心教授丘成桐与姚志刚为论文通讯作者。论文所有作者贡献相当。研究得到新加坡教育部研究基金和清华大学丘成桐数学科学中心的支持。
论文链接:
www.pnas.org/doi/10.1073/pnas.2400002121
供稿:数学科学中心
题图设计:任帅
编辑:李华山
审核:郭玲